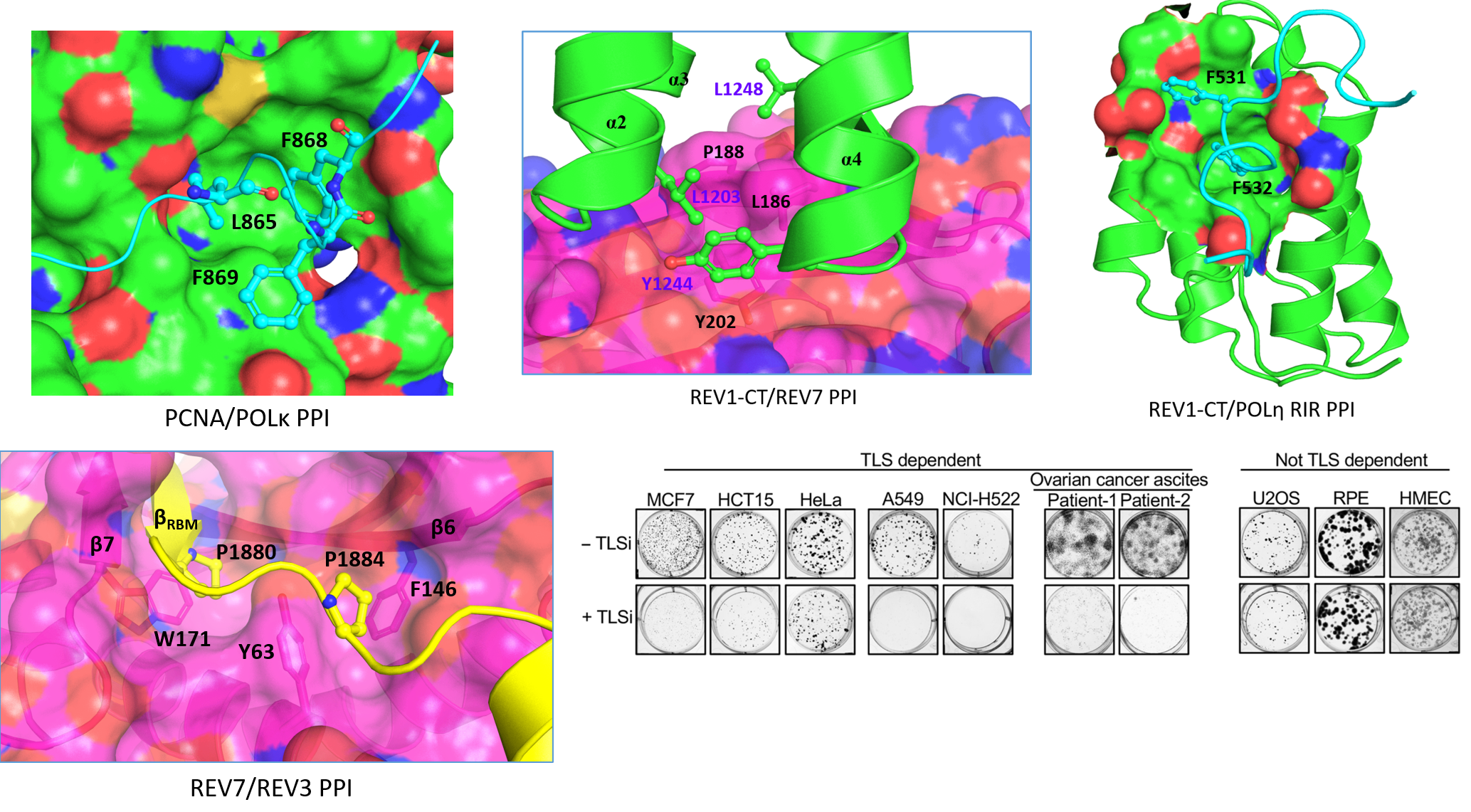
Translesion synthesis (TLS) is a DNA damage tolerance mechanism that replaces the replicative DNA polymerase with a specialized, low-fidelity TLS DNA polymerase that can copy past DNA lesions during active replication. Recent studies have demonstrated a primary role for TLS in replicating past DNA lesions induced by first-line genotoxic agents, resulting in decreased efficacy and acquired chemoresistance. With this in mind, targeting TLS as a combination strategy with first-line genotoxic agents has emerged as a promising approach to develop a new class of anti-cancer adjuvant agents.
The replicative bypass of bulky DNA adducts caused by these first-line agents is mediated by a set of specialized low-fidelity TLS DNA polymerases that can copy over DNA lesions while temporarily leaving them unrepaired. Multi-protein complexes that act in this process are comprised of the Y-family DNA polymerases Rev1, polη, polι and/or polκ and the B-family polymerase polζ assembled on a DNA-sliding clamp PCNA. Because TLS is inherently mutagenic, cells have evolved a complicated network of PPIs that regulate TLS function to ensure that the TLS heteroprotein complex is only recruited to sites of DNA damage. More specifically, these PPIs fine-tune assembly of the TLS complex, they localize the proper TLS DNA polymerase to the lesion site, and they regulate polymerase switching events between replicative and TLS polymerases. These PPIs also allow the TLS complex to adopt different configurations depending on the type of DNA lesion, where the lesion occurs, and what specific step in TLS is currently underway. The Hadden lab is utilizing multiple approaches to identify small molecules that disrupt several key TLS PPIs and develop them as anti-cancer agents.