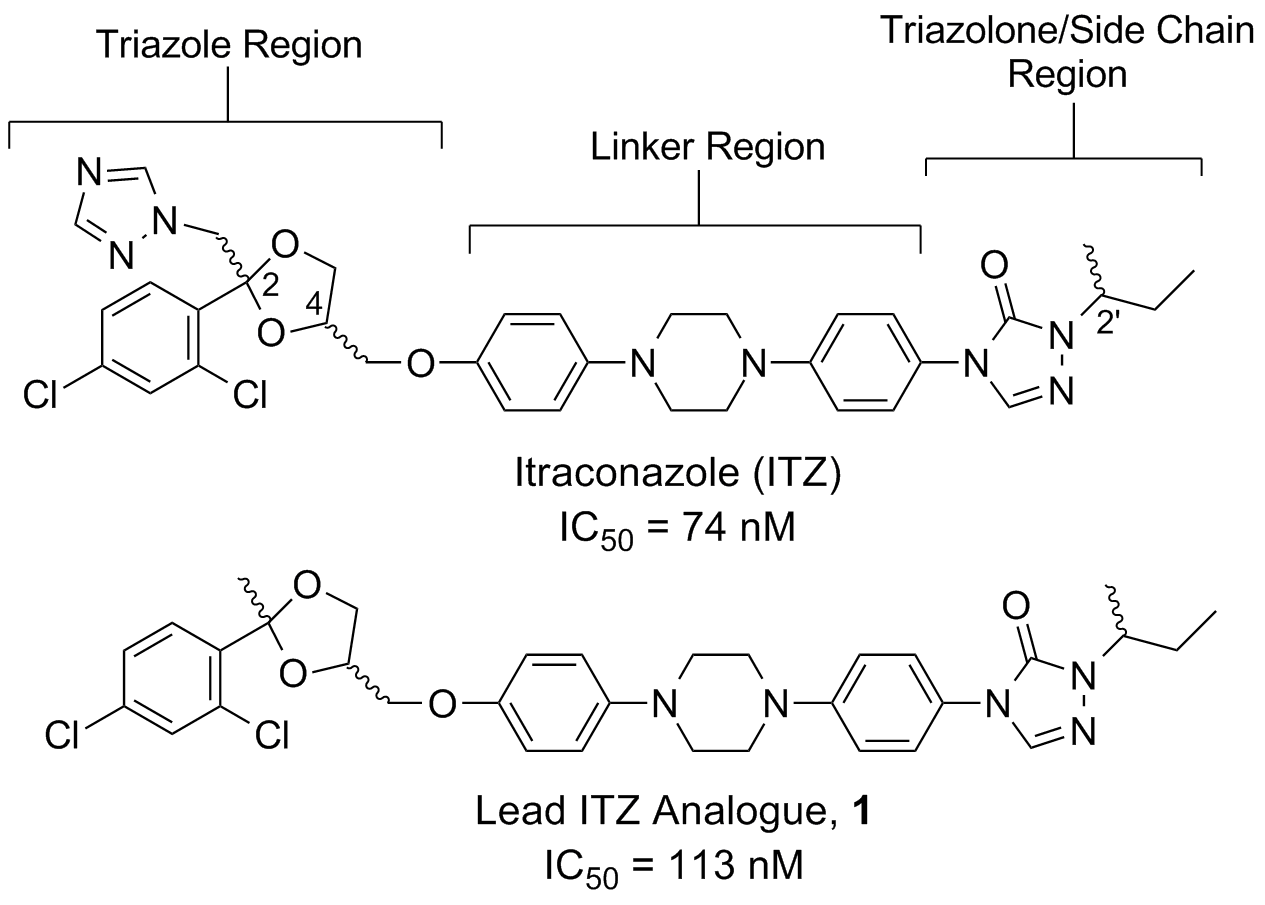
Itraconazole (ITZ) is a member of the triazole class of antifungal agents that exerts its antimycotic effects through potent inhibition of lanosterol 14α-demethylase (14LDM, also known as CYP51), a cytochrome P450 enzyme that catalyzes the oxidative conversion of lanosterol to ergosterol. The N4 of the ITZ triazole moiety interacts with the heme group in 14LDM, preventing coordination of the molecular oxygen required to initiate oxidation. This ability to coordinate the heme of CYP450s is also responsible for its most common detrimental side effect, inhibition of CYP3A4. CYP3A4 inhibition results in multiple drug-drug interactions and contraindications and requires careful monitoring of diet and drug regimens when ITZ is administered.
Recent studies have shown that ITZ also functions as a potent inhibitor of both Hh signaling and angiogenesis in vitro and in vivo (IC50 values ~100-700 nM, depending on assay and cell type). Most importantly, ITZ maintains inhibitory activity against multiple forms of Smo (including D473H) that confer resistance to GDC-0449 and prolongs survival of mice with GDC-0449 resistant forms of medulloblastoma. Preliminary structure-activity relationship (SAR) studies for ITZ-mediated Hh inhibition and angiogenesis performed in the Hadden lab have identified multiple regions of the ITZ scaffold that are amenable to modification. Our data suggests that further modifications to these regions will provide ITZ analogues that maintain potent Hh inhibition, while also demonstrating improved pharmacokinetic (PK) parameters and reduced off-target side effects characteristic of ITZ treatment.